Source : RechercheIndépendante.blogspot.com
Alexander Samuel est docteur en biologie moléculaire et auteur de plusieurs publications en lien avec le neurodéveloppement.
Titre original de l’article : Sars-cov-2 : Arme biologique ?
1) Un peu de contexte
La piste des virus a été étudiée pour fabriquer des armes biologiques. Par exemple, dès 1959 à la conférence de Pugwash sur les armes biologiques et chimiques, il a été question du virus arbor et de la petite variole. Les virus affectant les voies respiratoires ont été déjà à cette époque écartés comme étant de mauvais candidats. Mais en 1962, Geoffrey Bacon, un scientifique travaillant pour une base militaire secrète est mort d’une peste pneumonique.
Dans les années 1950, la myxomatose a été utilisée en Australie sur les lapins, y causant une forte mortalité. Cependant, les formes les plus bénignes ont été sélectionnées car elles permettaient de transmettre la maladie avant la mort de l’animal… Fabriquer un virus fortement pathogène ne va pas, à terme, être très efficace. Et justement la mortalité du sars-cov-2 est plutôt faible !
Quel est le but recherché dans une arme biologique ? Les militaires ont été décrits par les scientifiques les plus critiques comme ayant des idées très simples. On peut citer :
« En 1915, un chimiste anglais proposa au général en charge de ces questions l’utilisation de dichlorethyl sulfide. Le général demanda « Est-ce que c’est mortel? ». « Non » répondit le chimiste, « Mais cela peut incapaciter temporairement un énorme nombre d’adversaires d’un seul coup ». L’homme de sang répondit alors « Ce n’est pas bon pour nous, nous voulons quelque chose qui peut tuer ». Il est intéressant de noter à quel point les idées de ce valeureux soldat sont en totale adéquation avec celles d’un enfant de 5 ans d’intelligence moyenne »
– J.B.S. Haldane dans Callinicus : a Defence of Chemical Warfare
La logique militaire est très pragmatique en termes d’armes chimiques et biologiques. Elle souhaite un effet immédiat, très contrôlable, et déployable rapidement et facilement. Les virus n’ont aucun de ces avantages par rapports à des armes chimiques, ou des toxines. On ne maîtrise pas le temps d’incubation et la date à laquelle le virus fera un réel effet. Les virus demandent également d’être peu mortels pour se propager, ou alors il faut qu’ils aient un temps d’incubation très long permettant leur dissémination. Ces paramètres les rendent inopérants d’un point de vue militaire. Ils sont de plus incontrôlables et peuvent réinfecter les soldats du fait de mutations dans la population ciblée.
En termes d’arme biologique, les militaires ont toujours préféré les toxines et les produits chimiques, plutôt que les virus. Cependant, avec les progrès de la génétique, il est si facile de manipuler un génome qu’il est important de se prémunir d’un terrorisme biologique. C’est pour cela qu’effectivement, il existe des recherches un peu secrètes qui ont eu lieu. Peu d’informations ne filtrent, mais parfois on voit certains témoignages intéressants. le centre du Bouchet en est un bon exemple :
« Pour mieux répondre aux menaces grandissantes dans le domaine biologique, la Défense a décidé d’investir dans deux laboratoires qui sont complémentaires, permettant de manipuler des virus vivants de classe 4. Des virus contre lesquels il n’existe encore aucun traitement connu »
M. Le Drian à DGA Maîtrise NRBC (Nucléaire, radiologique, bactériologique, chimique)
Des fuites de virus ont déjà eu lieu par le passé et ont rapidement été identifiées, notamment un virus H1N1 fabriqué en 1950, oublié dans un frigo et qui a créé une épidémie de grippe en 1977 en URSS, ou encore des accidents de laboratoires chinois travaillant justement sur le sars-cov en 2004.
Dès lors, il paraît peu probable qu’une arme biologique développée intentionnellement se soit échappée d’un laboratoire. D’autant plus que le lieu de déploiement d’une arme biologique peut être choisi, et qu’on ne l’aurait pas libérée juste à côté du laboratoire P4 où le virus aurait été « développé ».
Reste alors une seule hypothèse possible. Dans le cadre de la recherche visant à se prémunir d’une pandémie future, soit par accident de type zoonose (transmission d’un animal à l’homme) soit par terrorisme biologique, il est possible en effet qu’un virus soit sorti d’un laboratoire par accident. Cependant, possible ne signifie en rien prouvé. Alors comment savoir, comment vérifier ?
2) Les bases en génétique
Il faut pour cela maîtriser un minimum la génétique et l’évolution. Nous avons tous une vague idée de l’ADN, certains se souviennent peut-être qu’il s’agit d’un grand livre de 6 milliards de bases, ces bases étant un alphabet à 4 lettres : A T G et C. Mais une grande partie de l’ADN est non codant. Les gènes ne représentent qu’une petite partie du livre. Le reste est fait de régions permettant un repliement tridimensionnel de la molécule d’ADN, une régulation des gènes par dépliement et repliement rendant accessible ou non le gène dans tel ou tel contexte. On a aussi émis l’hypothèse du « junk DNA« , qu’une partie de l’ADN ne sert vraiment à rien, il y a des reliques d’insertions multiples, de déplacements et de croisements de gènes qui ont disparu au cours de l’évolution…
La partie réellement fonctionnelle du génome étant relativement faible (1 à 2% du génome représente des parties réellement codantes), une mutation a souvent très peu d’impact sur le fonctionnement d’un organisme. Ainsi, les mutations ne sont pas rares et sont permises dans un être vivant, sans que cela n’ait de conséquences sur son fonctionnement.
On peut également rappeler le code génétique : 3 lettres correspondent à un acide aminé.

Mais une mutation par exemple de CUU transformé en CUC n’aura donc aucun effet puisque les deux codons sont lus comme étant ceux d’une « leucine ». Donc même en touchant les régions codantes (1 à 2% du génome), il y a de fortes chances qu’une mutation n’aie aucun effet. Il est intéressant parfois de ne pas comparer la séquence génétique, mais la séquence peptidique, c’est à dire la séquence en acides aminés, ces 20 briques permettant de fabriquer les protéines, déduites du code génétique. Chaque acide aminé a une lettre correspondante :
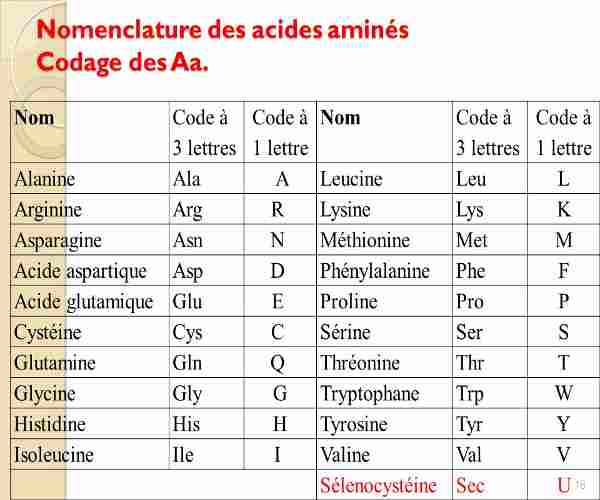
Quand une mutation a un effet majeur, touchant un gène essentiel, la cellule meurt et ne reproduira pas cette mutation. Mais elle peut aussi avoir un effet extrêmement mineur, ne changeant en rien la fonction d’une protéine. En réalité, la probabilité d’avoir une mutation ayant un sens est plutôt rare donc, sur un génome d’un être vivant comme chez l’homme.
Comment apparaissent les mutations ? Tout simplement à la copie : les enzymes (polymérases) qui recopient l’ADN font des erreurs. Chez l’homme, on est en moyenne à 4,4 erreurs toutes les 100 000 bases. Ces erreurs de copie vont être corrigées, ce qui permet encore de réduire grandement les mutations qui apparaissent dans l’ADN.
Les régions codantes de l’ADN sont transcrites en ARN, qui sort du noyau et va être lu par le ribosome pour fabriquer une protéine. Les erreurs de l’ARN polymérase semblent plus fréquentes, on arrive à une erreur toutes les 10 000 bases environ sur l’ARN transcrit.
Un virus possède son propre système de réplication, et emploie cependant une partie de la machinerie cellulaire pour se répliquer (notamment les ribosomes). Le taux d’erreur de copie peut donc être plus faible ou plus important chez un virus, et va avoir un rôle pour déterminer son évolution.
De nos jours, nous disposons aussi d’outils comme le système « Crispr-Cas« , qui permettent avec une grande finesse et une grande précision d’éditer à volonté une séquence génétique, de modifier comme on veut un virus en insérant à l’endroit que l’on souhaite la séquence que l’on veut. Cependant, chaque insertion et chaque modification est une manipulation unique à effectuer, et il est plus commode d’insérer un gros fragment de synthèse que d’en insérer des milliers très courts.
Comment dès lors savoir si un virus a été fabriqué en laboratoire ou s’il a muté naturellement par des erreurs de copie accumulées?
3) Le séquençage, réel outil de traçage.
Identifier si le sars-cov-2 est un objet d’étude échappé de laboratoire ou s’il est apparu par évolution naturelle revient à vérifier si la séquence génétique a pu être « fabriquée » ou si elle est apparue par évolution. Pour ce faire, on dispose de séquences d’autres virus, et notamment des séquences de tous les variants du premier sars-cov qui a déclenché une épidémie en Chine en 2002/2003.
C’est dans le contexte de cette épidémie que les chercheurs ont compris que cette famille de coronavirus pouvait infecter l’homme, et qu’une nouvelle épidémie pouvait se déclencher. Des romans ont alors émergé, certains spécialistes ont exprimé leur inquiétude, parlant d’une bombe à retardement. Ce qui n’a pas manqué d’apparaître dans une autre région du globe, au Moyen-Orient, avec le mers-cov en 2012.
Et c’est pour cette raison qu’à proximité même de lieu d’émergence de nouvelles pathologies du fait de la proximité entre hommes et animaux, que des laboratoires séquencent le génome viral apparu chez l’homme, mais également celui des virus chez des animaux qu’ils collectent dans la nature, pour voir l’évolution des mutations naturelles, et les enregistrent dans une base de donnée publiquement accessible.
Certains, sentant un potentiel filon pour gagner de l’argent grâce à un vaccin contre un futur virus, vont même jusqu’à déposer des brevets sur des séquences trouvées dans la nature, pour pouvoir s’assurer d’être les seuls à isoler ce variant et pouvoir l’utiliser comme base d’étude pour un vaccin. C’est le cas du fameux EP1694829B1 issu d’une souche Vietnamienne (Hanoï). Comme c’est un peu compliqué de déposer un brevet sur quelque chose trouvé dans la nature, les « inventeurs » vont user d’artifices, disant que le brevet concerne la technique d’extraction et l’analyse du génome viral, pour maintenir leurs droits dessus. Ils pensaient sans doute que cette souche pourrait être à l’origine d’une future zoonose et voulaient développer un vaccin.
De ce fait, nous disposons, accessibles publiquement, de nombreuses séquences de coronavirus animaux que l’on peut comparer entre eux, et avec le nouveau sars-cov-2. Si le virus est apparu par évolution, nous devrions donc retrouver les différents variants dans les séquences génomiques, au fur et à mesure de l’évolution, donc au fur et à mesure de nouvelles découvertes de virus dans le règne animal. Si par contre, il s’agit d’une fabrication humaine, nous ne devrions plus trouver de séquences intermédiaires.
Pour résumer, voici les différents scénarios envisagés :

La question est donc de savoir s’il existe des mutations et des variants suffisamment proches les uns des autres pour expliquer une lente évolution du virus, ou si une mutation paraît trop suspecte car elle implique un trop grand bond. Les spécialistes du domaine utilisent pour cela un outil mathématique appelé la « distance génétique ». Cette distance génétique est rapportée au temps d’évolution : si une trop grande distance génétique apparaît par rapport à la durée réelle, on peut devenir soupçonneux.
Une approche globale rapide consiste donc à se demander si la distance génétique entre le sars-cov de 2002/2003 et le sars-cov-2 de 2019/2020 peut être parcourue en ces 17 ans. Et ce qui est utile, c’est qu’on ne pourra pas être biaisés par une envie de prouver un complot car la distance génétique faisait déjà l’objet d’études dès 2006. Le génome du virus fait 30 000 bases de long, et le taux de mutation a été estimé entre 1,05 et 1,26 mutations toutes les 1000 bases par site et par an. En 17 ans, l’évolution du virus semble a priori impossible dans une hypothèse de mutations par évolution. Mais en réalité, on n’est pas parti du sars-cov de 2002/2003 pour aller au sars-cov-2. Les deux souches ont sans doute un ancêtre commun. Il y a environ 75% d’homologie entre la séquence du sars-cov et celle du sars-cov-2, ce qui reste assez proche pour permettre une lente évolution d’un ancêtre vers l’un et vers l’autre. En 2002/2003, une branche évolutive a fait une zoonose, et en 2019/2020, c’est une autre branche évolutive. On peut même dater l’ancêtre commun, puisqu’il y a une différence de 7500 nucléotides entre les deux variants, et donc une distance de « 200 ans » environ, soit un ancêtre datant des années 1910 environ (92 ans vers le premier sars-cov, 109 ans pour le second sars-cov-2)
4) La publication indienne foireuse
Malheureusement, dans l’urgence et la panique, les journaux scientifique ont accepté de diffuser largement des preprints, des publications scientifiques non validées, avant de les vérifier. Une publication rejetée a été faite par une équipe indienne, affirmant que c’est très improrbable d’avoir les mutations qu’ils ont repéré dans la séquence du virus par simple évolution… Je propose donc de vérifier cette affirmation.
Que nous disent ces génies ? Eh bien ils n’ont pas aligné les séquences nucléotidiques, mais les séquences peptidiques. C’est à dire les séquences en acides aminés. Voici ce qu’ils obtiennent entre sars-cov et sars-cov-2 :
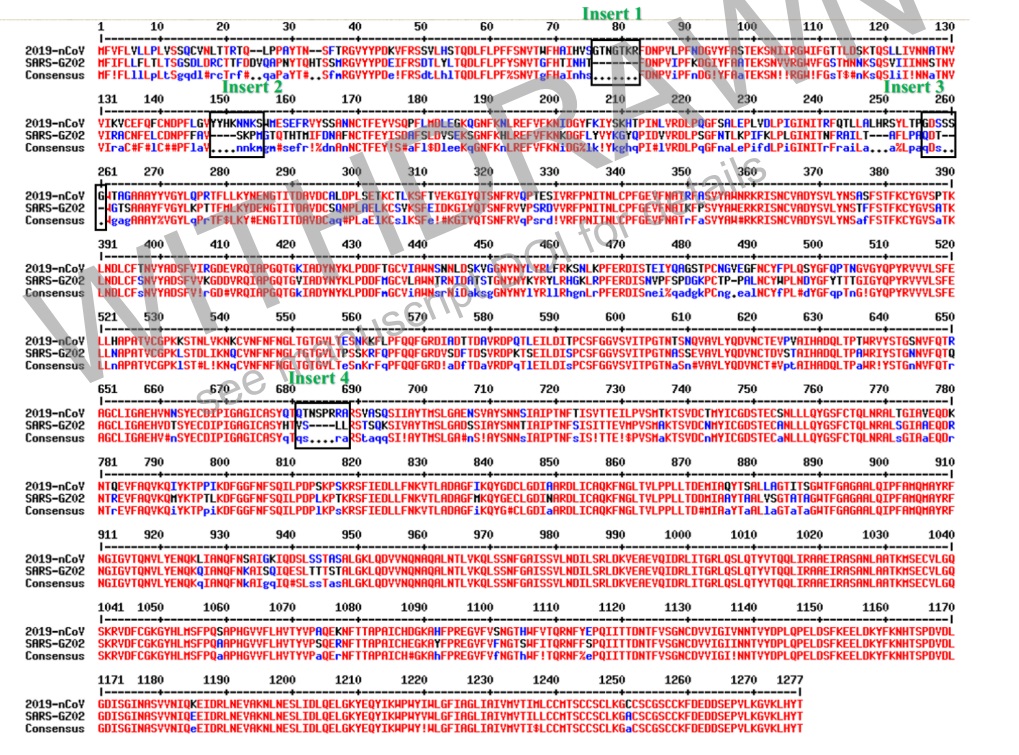
Les insertions aléatoires dans le génome existent, il se peut que, par la présence d’un autre ARN au même moment, un bout d’un ARN s’insère dans l’autre, ce sont des accidents rares mais possibles en 17 ans sur de nombreux organismes. La longueur de l’insert est importante dans un premier temps. En effet, une séquence insérée peut ressembler à autre chose par hasard, ou être suspecte.
L’insert 1 par exemple est GTNGTKR. Nous avons 20 acides aminés, donc grosso modo une chance sur 20 qu’un acide aminé inséré soit une G. Avoir GT revient à 1/20 x 1/20. GTN revient à 1/20 x 1/20 x 1/20 etc… On va donc mettre 1/20 à la puissance de la longueur de l’insert. Ici l’insert fait 7 acides aminés. 1/20 puissance 7 : soit une chance sur 1,28 milliards de tomber par hasard exactement sur la même séquence.
Cette probabilité paraît faible, mais je rappelle que nous avons séquencé des virus et des êtres vivants par milliards. Et une séquence d’un génome humain, je le rappelle, contient 3 milliards de paires de nucléotides, soit 1 milliard de « codons » de 3 nucléotides. Donc on est presque certains de retrouver une fois cette séquence GTNGTKR par hasard quelque part si on lit le génome humain.
Fort heureusement, un outil a été développé, le blastp. Pour le bien de la science, je vais vous demander de ne l’utiliser que si vous trouvez cela absolument nécessaire : les scientifiques du monde entier s’en servent, c’est un super calculateur qui compare des milliards de séquences, et chaque requête prend du temps à être traitée. Si tout le monde se met à blaster, on va faire ramer les machines et empêcher les scientifiques de travailler. Donc à moins de ne pas me faire confiance, merci de croire que j’ai blasté sincèrement. Voici le résultat :
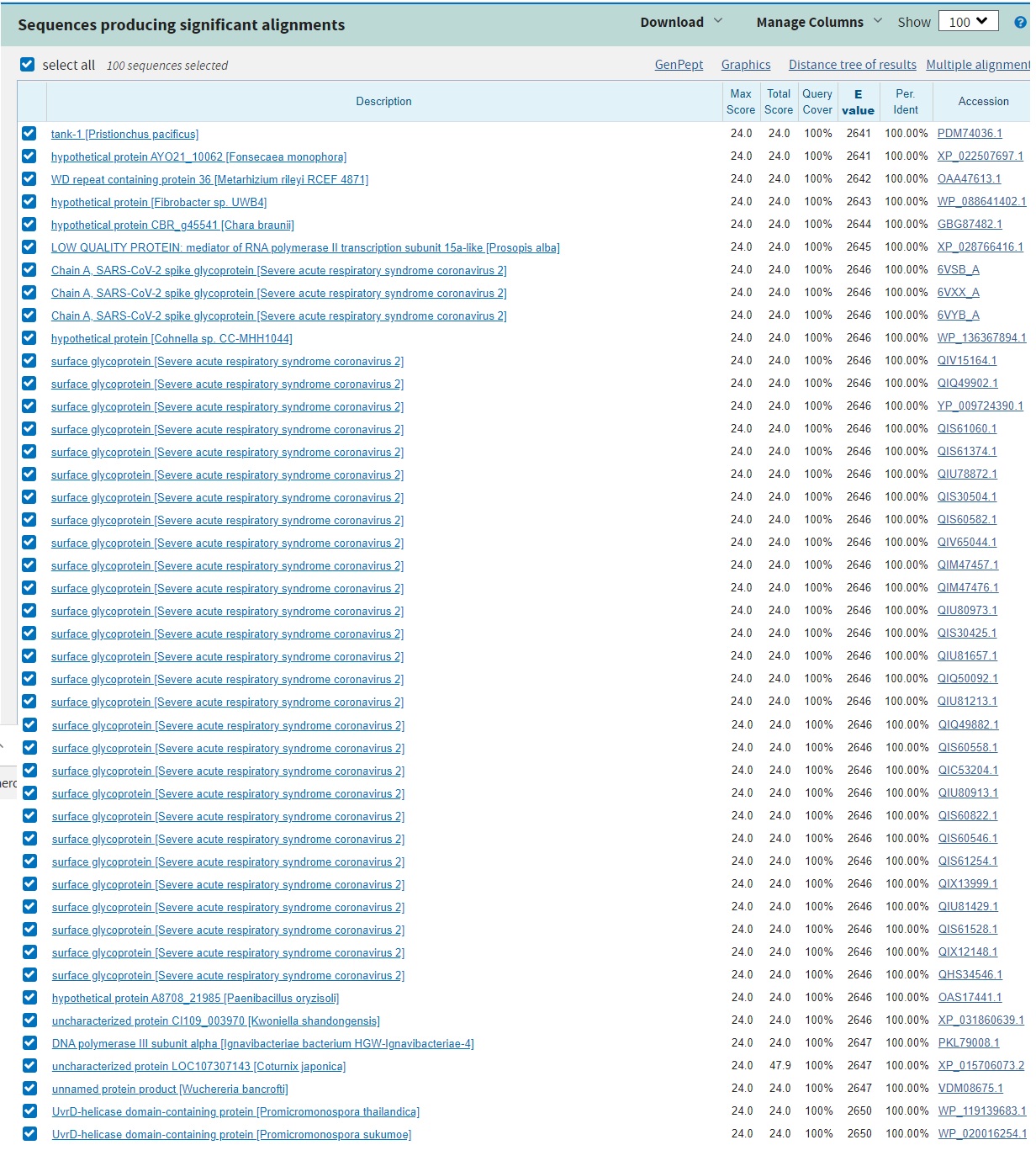
Outre le fait qu’il y a un alignement avec beaucoup de séquences de sars-cov-2, on voit apparaître une grande quantité d’autres homologies chez de nombreux autres virus et bactéries. Le nom est indiqué entre parenthèses.
Si on regarde en détail, on trouve d’ailleurs ce même insert chez une séquence virale de chauve-souris de 2013 appelée RaTG13 :

Le second insert est plutôt un artefact, quand on regarde la séquence, puisqu’on ne sait pas vraiment ce qui est inséré, toute la zone a beaucoup évolué et n’était sans doute pas soumise à une forte pression de sélection. On retrouve pourtant cette même séquence chez RaTG13 :

Le troisième insert est également présent depuis 2013 mais on le trouve dans d’autres séquençages aussi :

Finalement, la seule nouveauté du sars-cov-2 est une minuscule séquence PRRA qui n’est pas trouvée dans le règne animal :
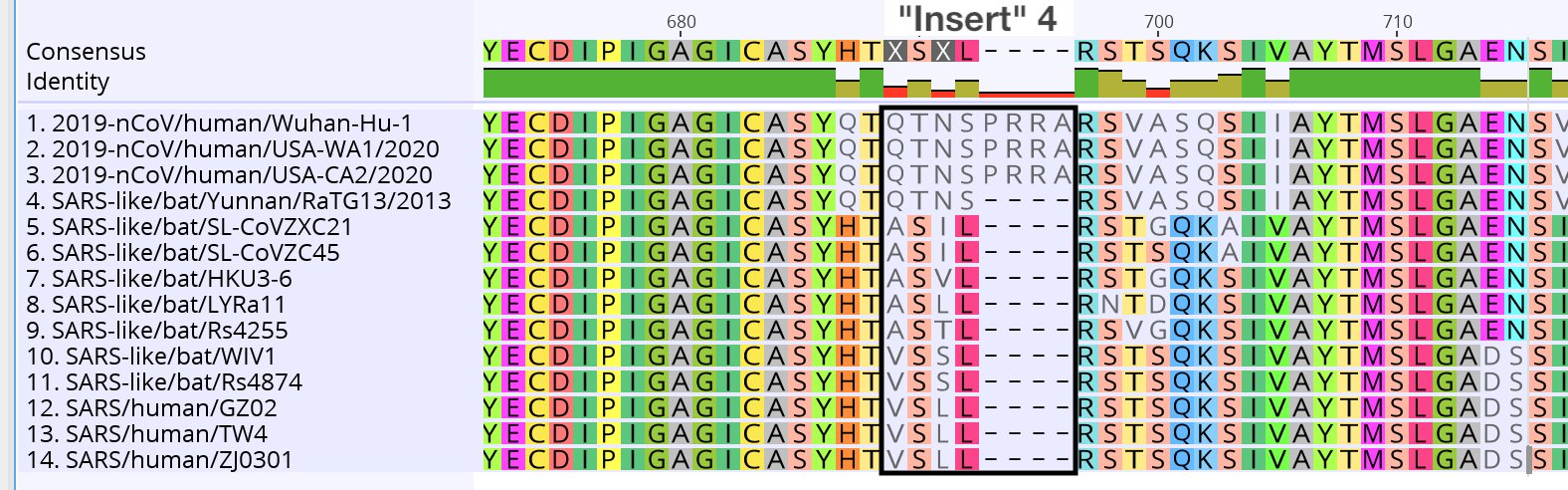
Pour les plus courageux, il existe des logiciels de lecture de séquence (format de fichier FASTA) comme UGENE (gratuit) qui permettent de regarder tout ça. Et très généreusement Trevor Bedford nous a proposé ces séquences déjà alignées avec blastp au format fasta, à télécharger ici.
Ce que les indiens ont fait remarquer, c’est que ces trois inserts s’alignaient plutôt bien avec des parties d’une protéine du virus du sida, gp120 :

Première remarque : ces régions sont trouvées un peu aléatoirement sur des séquences du virus HIV très différentes les unes des autres (Kenya, Thailand, India), sur des variants différents. Le quatrième insert s’aligne avec une autre protéine (Gag). On a donc grandement élargi le spectre des séquences sur lesquelles on a cherché un alignement. Et surtout, chose étonnante : jamais l’équipe indienne n’est allée regarder au niveau de la séquence génétique : celle-ci apporterait bien davantage de divergence entre les séquences HIV et la séquence sars-cov-2.
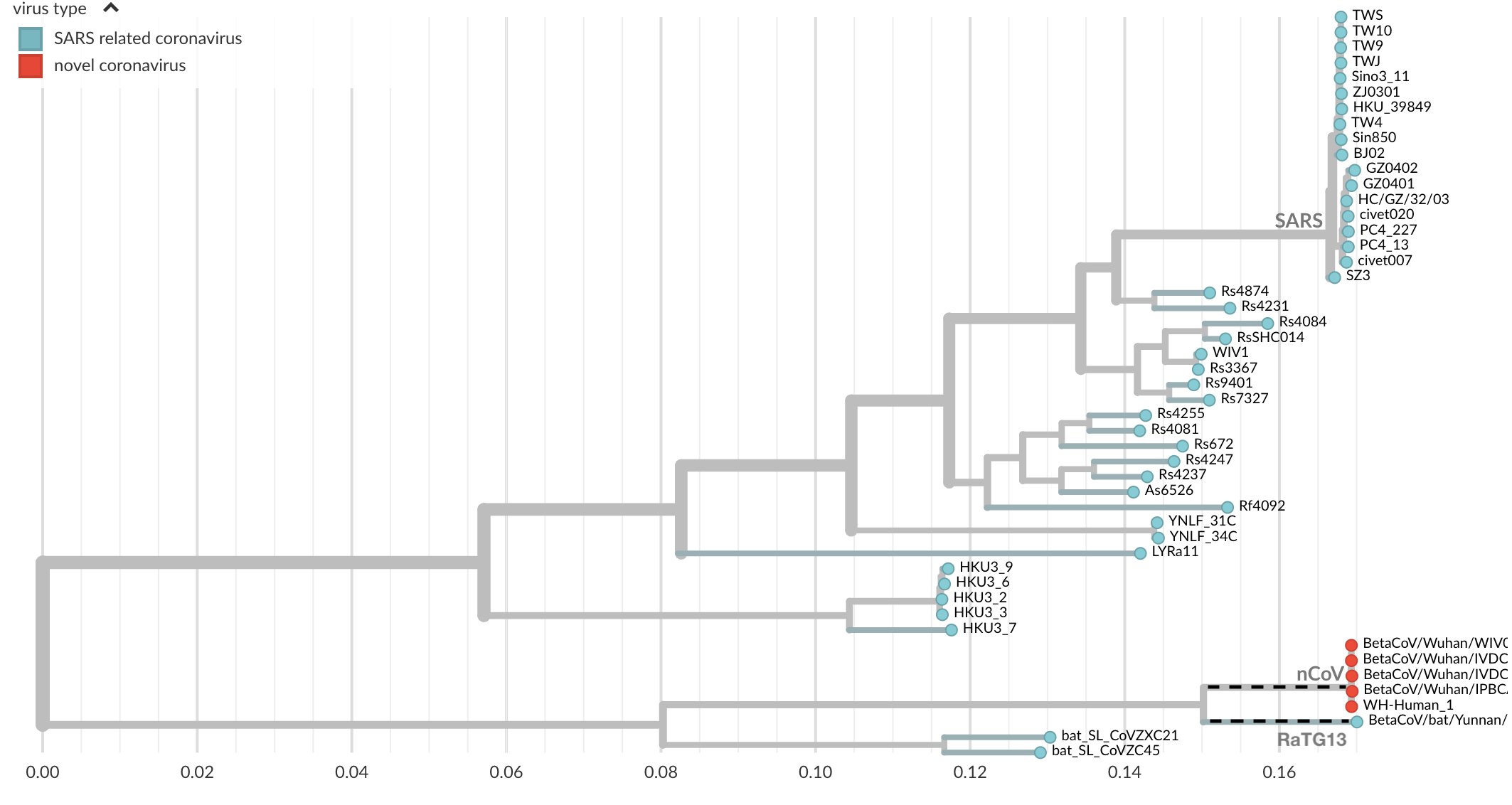
On voit donc une proximité énorme avec la séquence de RaTG13 qui n’a que 1100 différences avec sars-cov-2. Cette différence peut être représentée par un arbre dont les branches sont proportionnelles à la distance entre deux séquences. En pointillets, on voit la distance entre les deux.
La seule réelle nouveauté du virus introduit tout de même un site RRAR qui permet un clivage par la furine, donc une nouvelle fonctionnalité. Alors on peut poser la question de savoir si cette séquence précise n’a pas été ajoutée à la souche de chauve-souris RaTG13 de 2013, qui est très similaire au sars-cov-2. La séquence insérée est une séquence plus riche en bases G et C qu’en A et T, ce qui n’est pas très habituel dans l’ingénieurie génétique. Et si on prend l’ensemble de la séquence de cette zone, non plus au niveau de la protéine, mais au niveau de la séquence génétique, on trouve ceci :
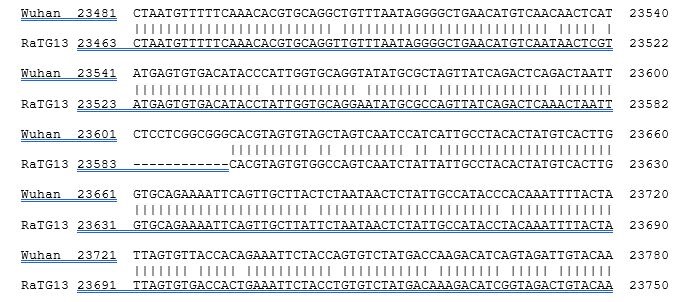
Il y a énormément de petites différences un peu partout. Il semble donc que RaTG13 ne soit en fait pas un ANCÊTRE, l’origine du sars-cov-2 comme on pouvait le penser de prime abord. Il semble plutôt que RaTG13 aie conservé les caractéristiques d’un ancêtre commun avec le sars-cov-2. Nous n’avons pas analysé l’intégralité de tous les virus présents dans tous les animaux, mais celui qui est le plus proche en terme de séquence (RaTG13) a accumulé des mutations sur son code génétique, qui n’ont pas affecté la séquence en acides aminés. Il est donc probable que le sars-cov-2 dérive d’un ancêtre qu’il a en commun avec RaTG13.
De quand date cet ancêtre ? D’après le taux de mutation (1/1000 à 3/10 000 bases par an), il devrait dater d’entre les années 1950 et 2000. On va créer un ancêtre commun artificiel au milieu, qui aura donc une différence de 554 nucléotides avec le sars-cov-2. Ce dernier aura aussi une différence au niveau de la séquence protéique de 79 acides aminés. Seules 14,3% des mutations ont eu un effet sur la séquence en acides aminés. En laboratoire, on n’aurait modifié que ce qui fait sens, on n’aurait pas introduit autant de mutations intermédiaires. D’autant plus que dans la nature on a trouvé RaTG13 qui lui est très proche génétiquement. Ce ratio de 14,3% est proche de ce qui se passe chez d’autres coronavirus :
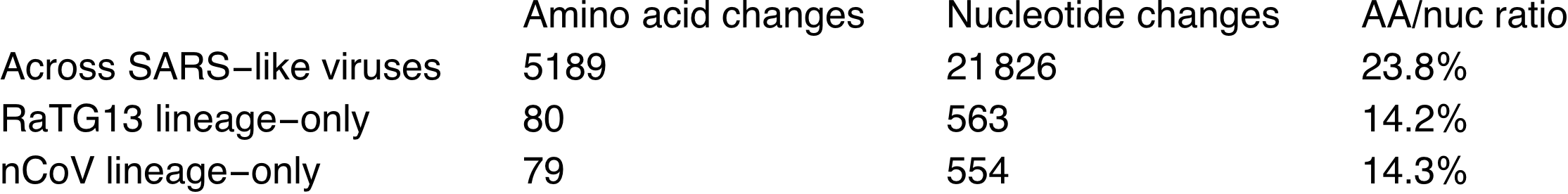
Un autre élément à vérifier est la localisation de ces mutations. En effet, si elles ciblent plus préférentiellement des éléments très utiles au virus, on peut supposer qu’il y a eu une intervention humaine. Voici la localisation des différences entre RaTG13 et les autres virus de la même famille :
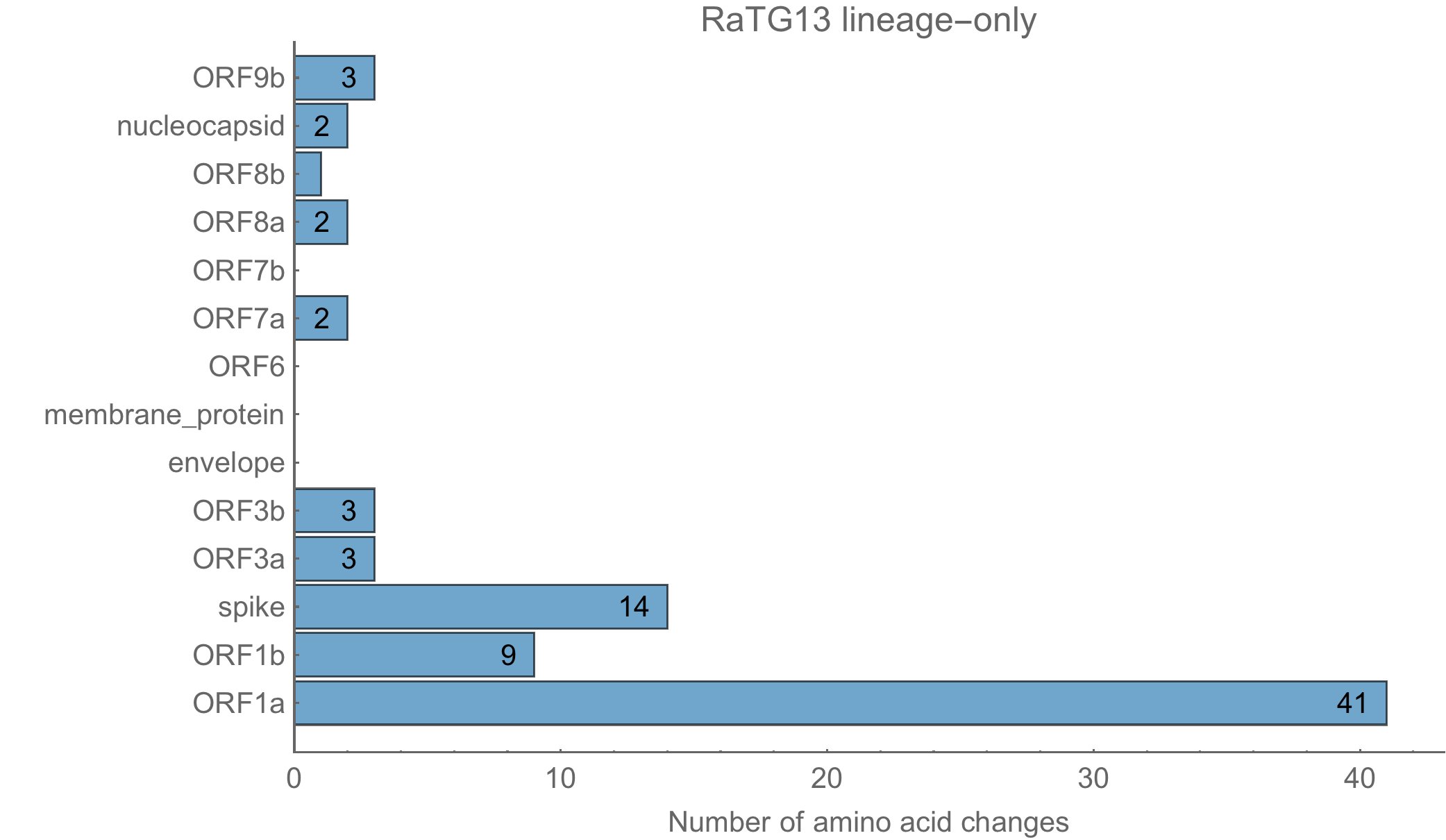
Et voici celles de sars-cov-2 :
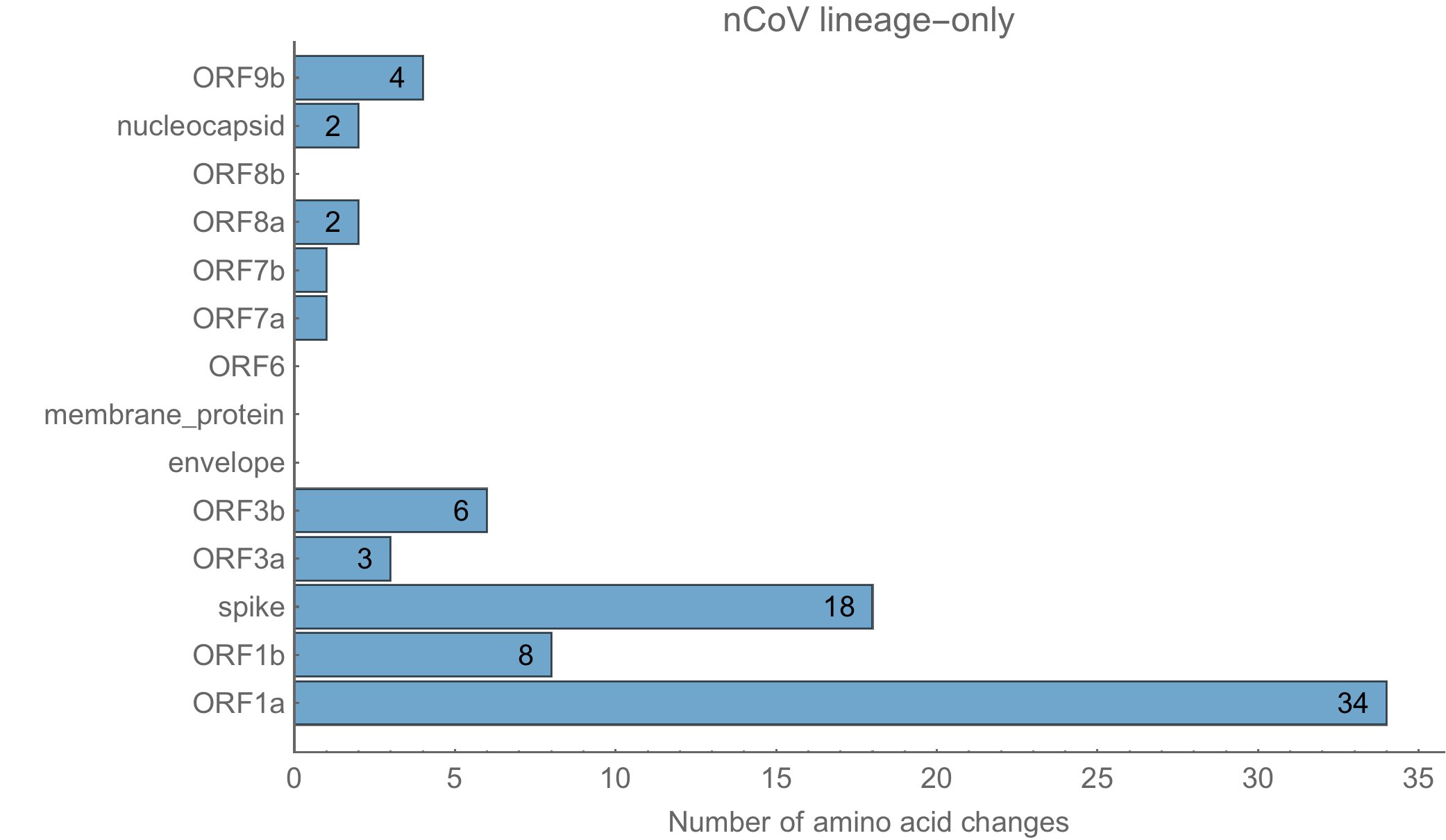
Les 79 à 80 acides aminés modifiés le sont sur diverses protéines, en respectant la même distribution chez sars-cov-2 que dans la souche naturelle RaTG13.
La généticienne Emma Hodcroft a proposé sur GitHub (pour les informaticiens) les outils permettant de reproduire soi-même toutes ces analyses.
5) Un peu de sérieux
Voici un travail plus sérieux réalisé par un spécialiste des séquences. on trouve en haut la séquence du sars-cov, et en bas la séquence du sars-cov-2. Au milieu, tous les intermédiaires rencontrés dans le règne animal. Ce schéma semble totalement correspondre à ce qui correspond à une évolution du virus abordée au point 3.
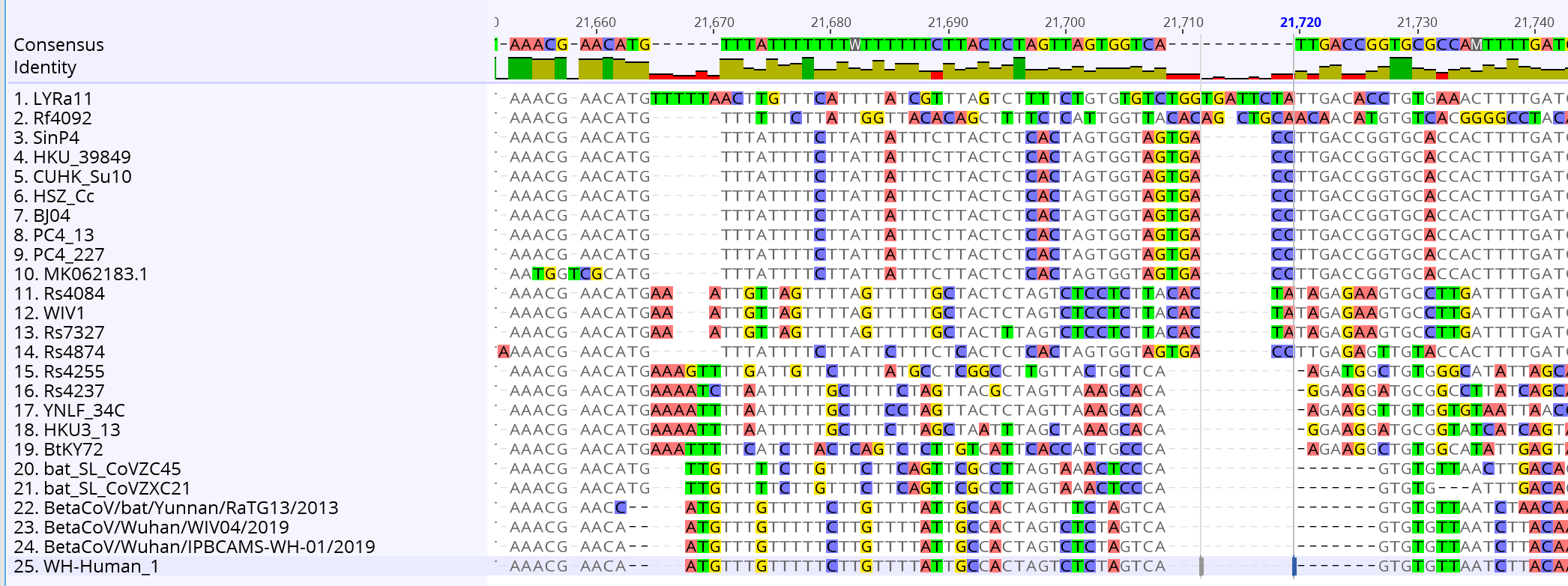
Nous voyons clairement des intermédiaires et une lente évolution. Pour prendre de la hauteur et ne pas regarder les mutations une par une, on utilise des logiciels de phylogénétique, calculant les distances génétiques entre deux séquences et construisant des arbres dont les branches sont proportionnelles à la distance entre deux séquences. Voici ce que cela donne si on regarde le premier sars-cov humain (rouge en haut), les chauve-souris, la civette et les séquences humaines su sars-cov-2 (ou ncov). On voit apparaître des intermédiaires entre les deux chez la chauve-souris, et ici on n’a même pas considéré tout le règne animal :
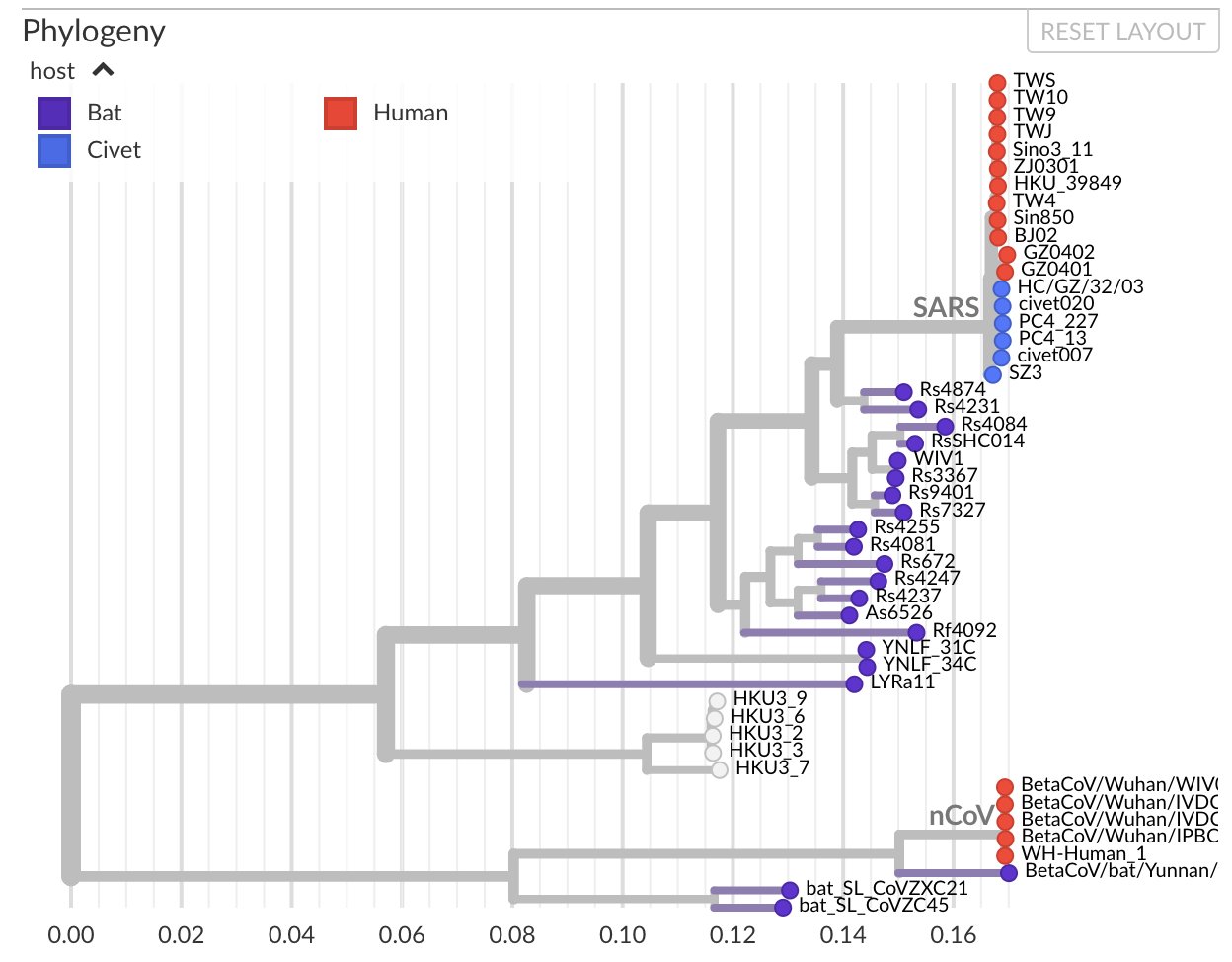
En regardant un peu plus largement, on peut aussi ajouter les séquences de pangolin très proches de celles de sars-cov-2, et regarder où se situe la souche de laboratoire la plus connue, WIV1 (flèche). On voit qu’elle est tout de même bien plus proche de sars-cov que de sars-cov-2, et bien en amont de nombreuses mutations animales rapprochant la séquence du sars-cov-2.
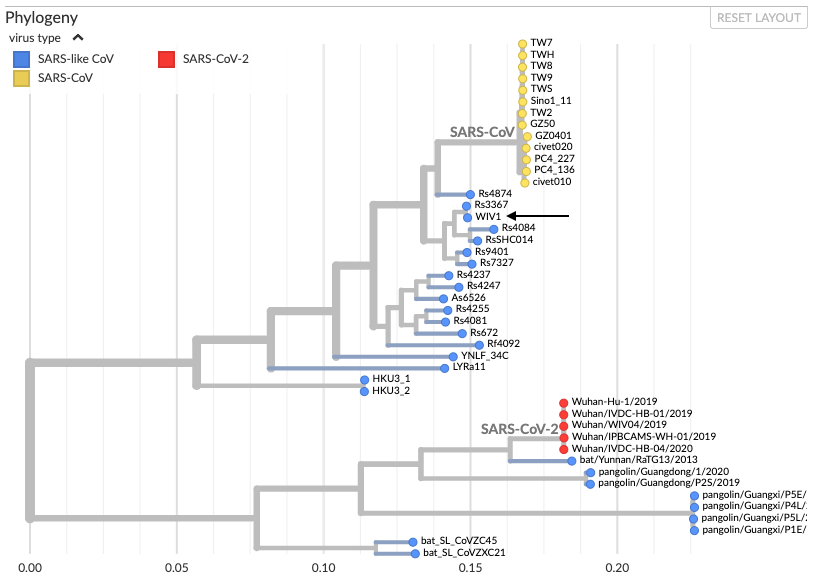
Si on retourne aux séquences, on trouve au total 6 mutations majeures permettant une meilleure liaison au récepteur ACE2 chez sars-cov-2. Ces mutations sont toutes retrouvées chez le pangolin.
Les arguments en faveur d’une origine animale, dite zoonose, s’accumulent d’autant plus que le marché de Wuhan a une forte proximité entre humains et animaux, et qu’on a retrouvé des séquences très concentrées dans l’environnement à l’Ouest du marché.


Comme j’aime beaucoup le travail scientifique de ce chercheur, je vais vous montrer ce qu’il a représenté à la vue de ces données :
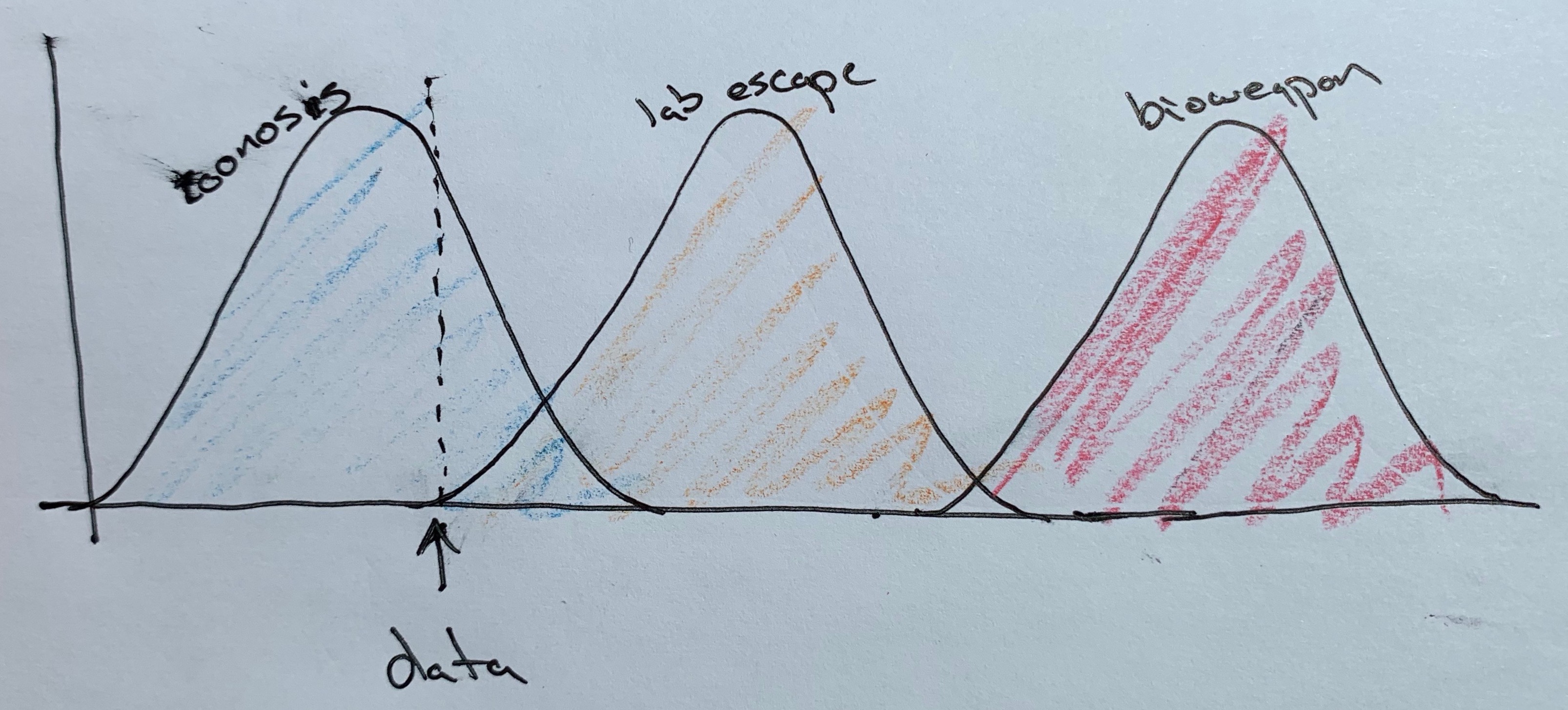
Ce sont les probabilités de zoonose (origine animale), accident de laboratoire ou arme biologique volontairement disséminée représentées graphiquement avec des gaussiennes, en termes de probabilités. On est dans la situation où la zoonose est très très hautement probable, et la fuite de laboratoire quasi impossible, d’après les données. L’hypothèse de l’arme biologique libérée volontairement devient infinitésimale.
6) Tant qu’on est dans les arbres foireux…
J’avais déjà critiqué un papier de la famille Forster. Je pensais qu’il serait vite enterré mais je vois que CNEWS vient de reprendre ce contenu complètement… foireux. Alors pourquoi foireux ? Eh bien parce que dans cette analyse, faite par une famille entre elle (on notera que tous les noms sont identiques et pour cause), qui ne savent pas se servir correctement des logiciels de phylogénétique, ils ont fait un arbre qui leur a indiqué 3 souches : A, B et C.
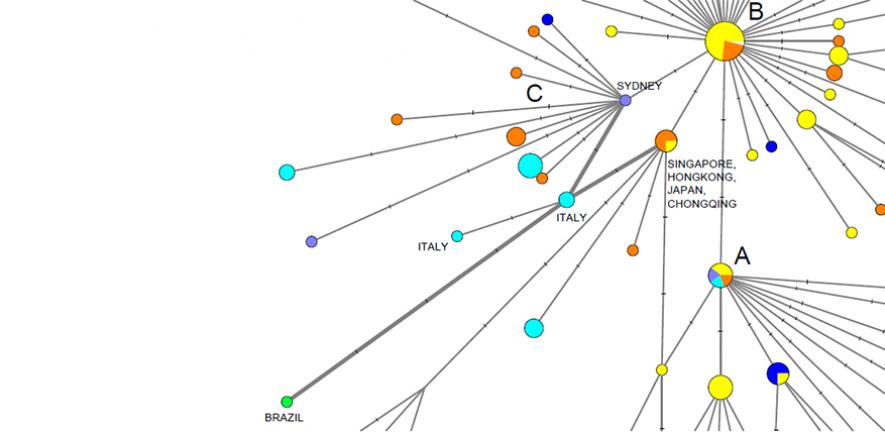
Or pour faire un tel arbre, on utilise la distance génétique entre toutes les souches séquencées rendues publiques sur GISAID. On les aligne par rapport à une séquence de référence, qui doit être un cousin, « outgroup » le moins lointain possible. Les spécialistes recommandent de ne pas dépasser 100 fois la différence intragroupe… Or ils ont outgroupé avec une séquence de chauve-souris 1100 fois plus éloignée, rendant toutes les analyses dans les groupes non significatives. Pour s’en rendre compte, voici à l’échelle l’outgroup :

Oui on ne voit pas grand chose, mais c’est normal. On verrait mieux si on n’avait pas outgroupé aussi loin… D’ailleurs, c’est à cause de cela qu’une initiative de reviewing rapide de papiers non publiés commence à être lancée. On en a grand besoin d’autant plus que forts de leurs résultats à trois souches complètement pétés, les Forster communiquent déjà dans la presse qu’ils ont trouvé une origine de l’épidémie en Septembre… Et si on se demande comment ce papier a pu être publié, eh bien… un des reviewers est à Cambridge Anthropology, comme les auteurs et l’autre reviewer est à Hambourg, université par laquelle Peter Forster est passé en 1997 pour passer sa thèse. Et Peter Forster étant un membre de l’Académie… La publication a sans doute été facilitée dans un grand journal comme PNAS.




Bonjour, je ne suis nullement compétente en génétique mais je vous remercie pour cet article aussi éclairant que compréhensible. Il me semble que l’hypothèse d’une erreur humaine est encouragée par deux informations « externes » dont la seconde est peut-être fausse. 1 ) Plusieurs lanceurs d’alerte chinois ont été réprimés. 2 ) la communauté internationale a salué la rapidité avec laquelle le génome sur Sars-cov2 a été séquencé par les Chinois. D’où une question subsidiaire : un labo peut-il avoir intérêt à rechercher dans la nature le plus grand nombre de virus « naturels », pour étudier leur potentiel épidémique et élaborer un vaccin… Lire la suite »
https://www.facebook.com/eric.gwizdek/posts/713729319456718